Directed evolution enzyme optimization is the process of accelerating natural selection in the laboratory to engineer enzymes with superior activity, stability, or substrate specificity across iterative mutation and screening cycles. Unlike rational design, it requires no prior structural knowledge of the target protein, making it the most broadly applicable protein engineering method available to researchers today. Recent campaigns have demonstrated over 24-fold improvements in enzyme activity and thermal resistance after just three rounds of evolution. Platforms like PACE (Phage-Assisted Continuous Evolution) and tools such as OptoPACE have further compressed timelines, while AI-assisted computational redesign now extends gains post-evolution. The result is a methodology that is faster, more accessible, and more powerful than at any previous point in biotech history.
How does directed evolution optimize enzymes?
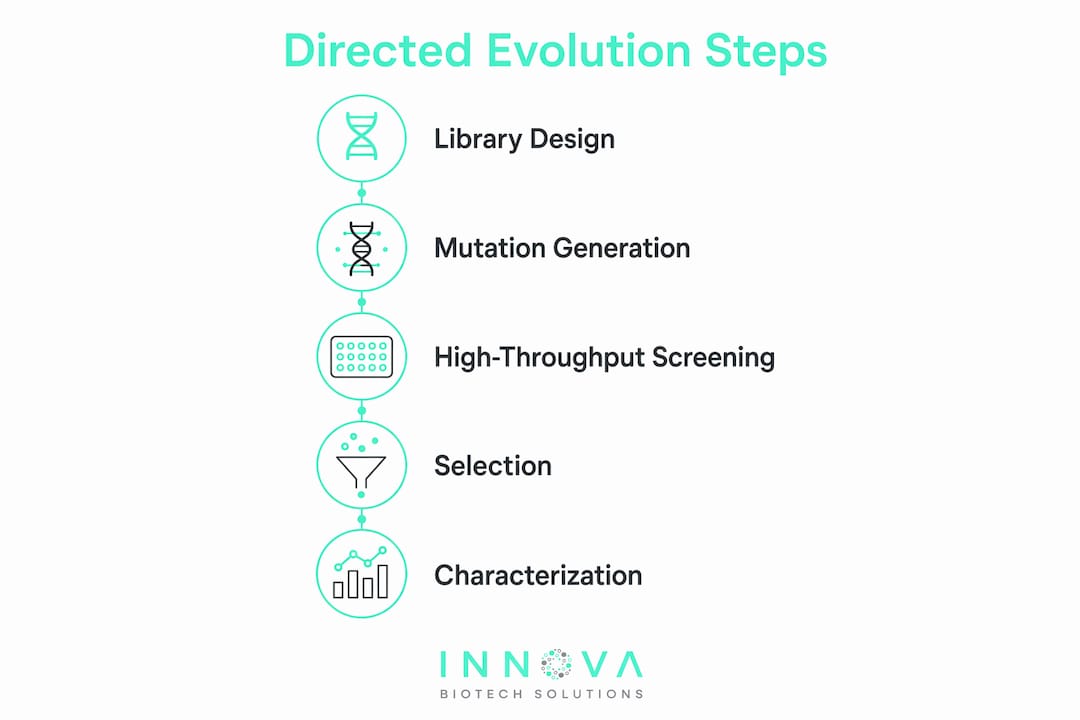
Directed evolution operates through two coupled steps: generating genetic diversity and selecting for improved function. The quality of your mutation library determines the ceiling of what selection can find, so library construction is where most campaigns succeed or fail.
The most common protein engineering methods for generating diversity include:
- Error-prone PCR (epPCR): Introduces random point mutations across the entire coding sequence at a tunable rate. Mutation frequency is controlled by adjusting Mn²⁺ concentration or dNTP imbalance. Typical targets are 1 to 3 amino acid substitutions per gene per round.
- DNA shuffling: Fragments homologous gene sequences and reassembles them via PCR, recombining beneficial mutations from multiple parent sequences simultaneously. This technique, pioneered by Willem Stemmer, is particularly effective when you have a family of related enzymes with known activity differences.
- Site-saturation mutagenesis (SSM): Targets specific residues for exhaustive substitution with all 20 amino acids. SSM is most efficient when structural or computational data narrows the search space to a handful of positions.
- Continuous evolution via PACE: Couples phage replication to the desired enzyme activity, allowing over 500 hours of uninterrupted directed evolution without manual intervention. The evolved RebH halogenase variant RebHEvo4 accumulated 12 mutations and showed a 5-fold increase in biosensor signal correlated with catalytic improvement.
The choice between in vivo and in vitro mutagenesis carries real trade-offs. In vivo platforms integrate mutation, translation, selection, and replication inside living cells, accelerating enzyme optimization without requiring structural data. In vitro methods offer tighter control over mutation rate and library composition but demand more manual handling between rounds. For most researchers running multi-round campaigns, in vivo platforms reduce labor per cycle significantly.
Pro Tip: Set your epPCR mutation rate to produce an average of 1 to 2 amino acid changes per variant in early rounds. Libraries with too many simultaneous mutations generate mostly nonfunctional sequences, which dilutes the useful signal in your screen.

What screening strategies identify the best enzyme variants?
High-throughput enzyme screening is the rate-limiting step in most directed evolution campaigns. A library of 10⁶ variants is only useful if your assay can process and rank them accurately at that scale.
Effective screening approaches fall into three categories:
- Functional activity assays: Colorimetric or fluorometric substrate turnover assays run in 96-well or 384-well plate formats. These are the workhorses of most labs and are best suited to enzymes with chromogenic or fluorogenic substrates.
- Biosensor-based screening: Metabolite-responsive transcription factors or riboswitches drive reporter gene expression (typically sfGFP) in proportion to product concentration. This format enables screening at the single-cell level via FACS, dramatically increasing throughput.
- Growth-coupled selection: Links enzyme activity to host cell survival, allowing selection across populations of 10⁸ or more. PACE is the most refined implementation of this principle.
One critical and frequently overlooked issue is assay calibration. Biosensor signals can saturate at high product concentrations, causing the genotype-to-phenotype mapping to become non-linear. In the 2026 PACE halogenase study, the sfGFP biosensor saturated at high 7-chlorotryptophan levels, meaning the real catalytic improvement was larger than the measured signal suggested. Researchers who do not validate the linear range of their assay will systematically underrank their best variants.
A second bottleneck that receives less attention is protein secretion. Secretion signal optimization alongside directed evolution improved screening throughput and variant selection quality in a 2025 ACS study on chitosanase. If your enzyme is secreted and your signal is weak, the problem may be upstream of catalysis entirely.
| Screening format | Throughput | Best application |
|---|---|---|
| Plate-based colorimetric assay | 10³ to 10⁴ variants/day | Enzymes with chromogenic substrates |
| FACS with biosensor circuit | 10⁶ to 10⁸ variants/day | Metabolite-producing enzymes |
| PACE continuous selection | Continuous, population-scale | Long-term activity and solubility evolution |
| Growth-coupled selection | 10⁸ variants/selection | Essential metabolic pathway enzymes |
Pro Tip: Before committing to a biosensor-based screen, generate a standard curve spanning the full expected product concentration range. If the signal plateaus below your predicted top-variant output, redesign the circuit with a lower-affinity transcription factor or reduce inducer concentration.
How do multi-mutation interactions shape evolution outcomes?
The intuitive model of directed evolution places beneficial mutations at or near the active site. Structural data consistently contradicts this. Enzymes evolved for thermostability via 7 to 13 mutations outside the active site reorganized active site geometry indirectly and improved melting temperature by 17°C. The mutations worked through allosteric networks, core packing rearrangements, and salt bridge formation, none of which are predictable from sequence alone.
This phenomenon, called epistasis, means that the fitness effect of mutation A depends on whether mutation B is already present. Practically, this has three implications for how you design your campaigns:
- Do not restrict libraries to active site residues. Mutations at loops, subunit interfaces, and hydrophobic cores frequently contribute more to stability gains than active site substitutions. Libraries designed around distal beneficial mutations capture synergistic effects that active-site-only designs miss entirely.
- Run multiple sequential rounds rather than one large library. Each round fixes beneficial mutations from the previous round, creating the genetic background in which the next layer of epistatic interactions can be discovered.
- Use structural and computational analysis between rounds. Mapping where mutations cluster after round 2 or 3 reveals the allosteric networks your enzyme is exploiting. This information directly informs where to focus site-saturation mutagenesis in subsequent rounds.
Post-evolution, computational redesign constrained by distance and conservation thresholds can improve global stability without disrupting the catalytic gains already achieved. A 2026 Nature Biotechnology study on reverse transcriptases demonstrated this approach, using distance-based cutoffs and conservation filters to stabilize evolved variants while preserving function. This two-phase strategy, directed evolution followed by computational stabilization, is becoming standard practice for industrial biocatalyst development.
What recent advances are making directed evolution more efficient?
The gap between what directed evolution can theoretically achieve and what a typical lab can practically execute has narrowed considerably since 2024. Several converging developments are responsible.
| Technology | What it enables | Practical impact |
|---|---|---|
| OptoPACE bioreactor | Continuous PACE under $200 in hardware | Removes cost barrier to long-term continuous evolution |
| AI-assisted redesign | Mutation prioritization and stability prediction post-evolution | Reduces experimental load in stabilization phase |
| Automated liquid handling | 384-well screening with robotic colony picking | Increases throughput 10x over manual plate workflows |
| Expression system engineering | Signal peptide optimization alongside evolution | Eliminates secretion bottlenecks that mask true catalytic gains |
| In vivo hypermutation platforms | Targeted mutagenesis of specific loci in living cells | Accelerates cycle times and reduces library preparation labor |
The OptoPACE device deserves specific attention because it addresses a real access problem. Continuous evolution hardware has historically required custom-built bioreactors costing thousands of dollars. OptoPACE operates with simple components, provides turbidostat functionality with real-time turbidity feedback, and supports multispectral stimulation for optogenetic control. For labs running PACE for the first time, this lowers the barrier from prohibitive to manageable.
On the computational side, machine learning models trained on fitness landscapes from previous evolution rounds can prioritize which variants to synthesize and test next. This is not a replacement for experimental screening. It is a filter that concentrates experimental effort on the highest-probability candidates, which matters when your synthesis budget is finite. Innovabiotech integrates virtual screening methods with experimental directed evolution workflows to help clients prioritize variants before committing to wet lab resources.
The trend toward integrated in vivo platforms is also accelerating. These systems couple mutation generation, selection, and amplification inside a single cellular host, compressing what previously required weeks of manual library preparation into continuous automated cycles. For researchers optimizing enzymes with complex fitness landscapes, this speed advantage compounds across rounds.
Key takeaways
Directed evolution enzyme optimization works best when iterative mutation, calibrated high-throughput screening, and post-evolution computational redesign are treated as a single integrated workflow rather than separate steps.
| Point | Details |
|---|---|
| Library design beyond active sites | Include distal and loop residues to capture epistatic and allosteric beneficial mutations. |
| Assay calibration is non-negotiable | Validate biosensor linearity across the full product concentration range before screening. |
| Secretion bottlenecks mask true gains | Optimize signal peptides alongside directed evolution for secreted enzyme campaigns. |
| Continuous evolution is now accessible | OptoPACE hardware brings PACE workflows within reach of standard research budgets. |
| Computational redesign extends gains | Apply distance-based conservation filters post-evolution to stabilize top variants without losing activity. |
Why most directed evolution campaigns underperform their potential
I have seen researchers invest months into a directed evolution campaign and walk away with marginal gains, not because the method failed, but because two avoidable mistakes compounded across rounds. The first is assay non-linearity. When your biosensor saturates at the output level your best variants produce, you are selecting the second tier of your library, not the first. I cannot overstate how often this happens and how rarely it gets caught before the campaign ends.
The second mistake is treating the active site as the only relevant search space. Every structural study I find compelling on this topic points to the same conclusion: the mutations that matter most are often the ones you would never have predicted from the crystal structure. Designing libraries that permit accumulation of distal mutations is not a theoretical nicety. It is the difference between a 3-fold improvement and a 24-fold improvement.
My practical advice is to run a calibration experiment before your first screening round, not after. Map your assay's linear range, confirm your expression system is not the bottleneck, and then design your library to cover the full protein sequence rather than just the binding pocket. If you are running continuous evolution, invest the time to understand your hardware setup. Culture density monitoring and induction timing directly influence selection pressure and therefore which variants survive. These are not details. They are the experiment.
The researchers who get the most out of directed evolution treat it as a systems problem, not a mutagenesis problem. The mutation is easy. Everything around it is where the work actually happens.
— Hooman
How Innovabiotech supports your enzyme optimization projects
Innovabiotech combines computational and experimental expertise to support directed evolution campaigns from library design through post-evolution stabilization.

Innovabiotech's enzyme optimization services cover the full workflow: mutation library design, high-throughput screening support, expression system engineering, and AI-assisted variant prioritization. For researchers integrating computational redesign with experimental evolution, the protein engineering and computational modeling team applies distance-based conservation filters and structural analysis to stabilize top variants without sacrificing catalytic gains. Whether you are running your first epPCR campaign or scaling a PACE workflow for industrial biocatalyst development, Innovabiotech's team works directly with you from initial design through final variant delivery.
FAQ
What is directed evolution in enzyme engineering?
Directed evolution is a protein engineering method that mimics natural selection in the laboratory by iteratively mutating an enzyme gene and screening the resulting variants for improved function. It requires no prior structural knowledge and can optimize activity, stability, substrate specificity, and solubility simultaneously.
How many rounds of directed evolution are typically needed?
Most successful campaigns require three to five rounds to achieve significant improvements. A 2026 MDPI study on Bacillus subtilis chitosanase achieved over 24-fold gains in activity and thermal resistance across three rounds, which is consistent with results seen in other well-designed campaigns.
What is the difference between PACE and standard directed evolution?
Standard directed evolution requires manual library preparation and screening between each round, typically taking days to weeks per cycle. PACE couples phage replication to enzyme activity, enabling continuous evolution for hundreds of hours without manual intervention, as demonstrated by the RebHEvo4 halogenase variant that accumulated 12 beneficial mutations over 500 hours.
Why do beneficial mutations appear outside the active site?
Enzymes function through dynamic conformational changes, and mutations at loops, hydrophobic cores, and subunit interfaces can reorganize active site geometry indirectly through allosteric networks. PNAS structural studies show that mutations improving thermostability by 17°C were located entirely outside the active site, acting through core packing and salt bridge rearrangements.
How does computational redesign complement directed evolution?
After directed evolution identifies high-performing variants, computational redesign applies conservation and distance-based filters to introduce stabilizing substitutions without disrupting catalytic residues. This post-evolution step, demonstrated in a 2026 Nature Biotechnology study on reverse transcriptases, extends the functional lifetime of evolved enzymes for industrial and therapeutic applications.