
Accurate peptide binding affinity prediction sits at the center of modern drug discovery and protein engineering. Yet most researchers hit the same wall: computational models that perform beautifully on benchmark datasets collapse when applied to novel peptide-protein pairs with limited structural data. The affinity ligand market is projected to reach $2.66B by 2030 at a 7.8% CAGR, which means the pressure to get predictions right, fast, has never been higher. This guide walks you through the data requirements, computational methods, and tiered workflows that actually hold up in practice.
Table of Contents
- Key takeaways
- Peptide binding affinity prediction: data requirements
- Computational approaches for predicting peptide interactions
- Building a tiered prediction workflow
- Interpreting results and validation strategies
- My perspective on where affinity prediction actually breaks down
- How Innovabiotech supports your prediction workflows
- FAQ
Key takeaways
| Point | Details |
|---|---|
| Data quality drives accuracy | Prediction reliability depends on curated sequence and structural data before any model is applied. |
| Method selection is context-dependent | Sequence-based models outperform structure-based ones when high-resolution structural data is unavailable. |
| Tiered workflows reduce false positives | Combining ML screening with physics-based refinement improves lead prioritization in drug discovery pipelines. |
| Uncertainty quantification matters | Bayesian approaches flag unreliable predictions and prevent costly false positives in virtual screening. |
| Validation is non-negotiable | Computational predictions must be cross-validated and benchmarked against experimental data before acting on results. |
Peptide binding affinity prediction: data requirements
Before you run a single prediction, your data foundation determines everything. Garbage in, garbage out applies here more literally than in almost any other computational biology context.
What experimental and computational data you need
You need two categories of input: sequence data and structural data. Sequence data includes the primary amino acid sequences of both the peptide and the target protein, ideally with known binding annotations from curated databases like PDBbind, BindingDB, or the Immune Epitope Database (IEDB) for MHC-related work. Structural data means high-resolution crystal structures or cryo-EM models, preferably with co-crystallized peptide ligands to capture binding-induced conformational changes.
The quality threshold matters as much as quantity. Structures resolved below 2.5 Å are generally reliable for docking-based workflows. Anything above 3.0 Å introduces enough positional uncertainty that physics-based scoring functions will produce noisy outputs.
Benchmark datasets and preprocessing tools
| Dataset | Coverage | Best use case |
|---|---|---|
| PDBbind | Protein-ligand complexes with Kd/Ki values | Structure-based affinity modeling |
| BindingDB | Broad small molecule and peptide data | ML model training and validation |
| IEDB | MHC-peptide binding data | Immunology and vaccine design |
| S34 benchmark | Curated peptide-protein test set | Method benchmarking and comparison |
For preprocessing, tools like RDKit handle molecular feature extraction, while HTMD and MDAnalysis support trajectory-level structural analysis. Feature engineering for sequence-based models typically involves encoding schemes like one-hot, BLOSUM62, or learned embeddings from protein language models such as ESM-2. Getting this step right is where most teams underinvest, and it shows in their downstream prediction variance.
Pro Tip: Always split your dataset by sequence identity below 30% when building train/test sets. Models trained and tested on high-similarity sequences produce inflated performance metrics that will not generalize to novel targets.
Computational approaches for predicting peptide interactions
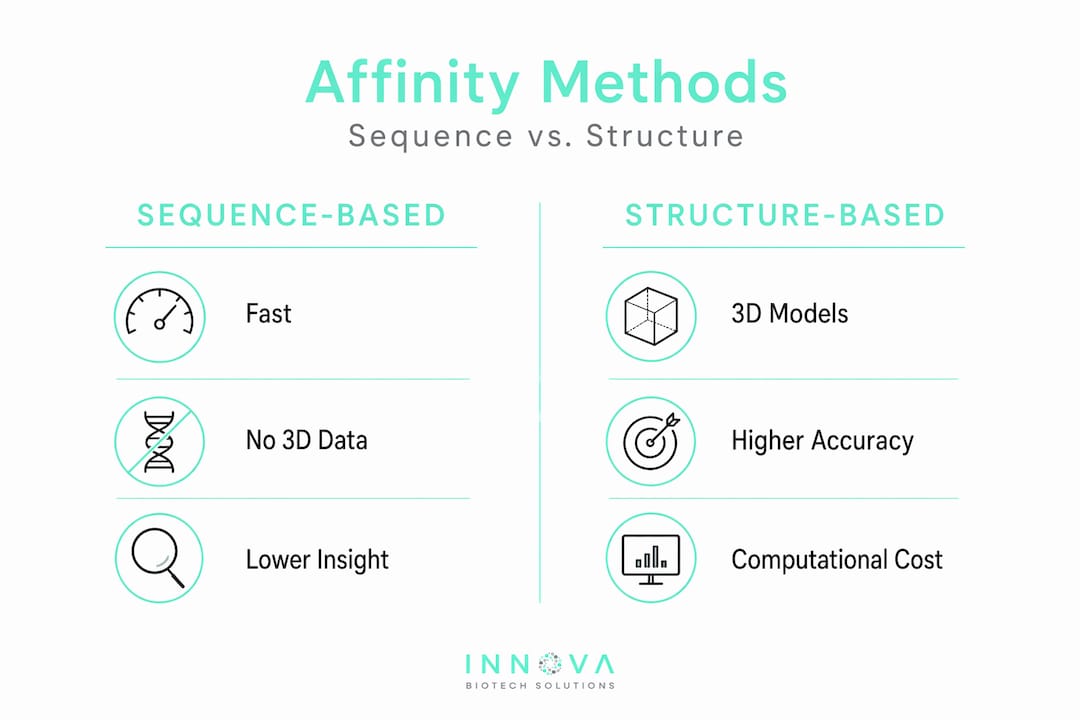
Three major classes of methods dominate binding affinity modeling: sequence-based, structure-based, and hybrid deep learning frameworks. Each has a specific niche, and choosing the wrong one for your data context is one of the most common and costly mistakes in the field.
Sequence-based models
Sequence-based approaches work directly from amino acid sequences without requiring 3D structural inputs. This makes them the go-to option for high-throughput screening scenarios where structural data is sparse or unavailable. Models like CrossAffinity use attention mechanisms and learned residue-level representations to capture binding-relevant patterns. CrossAffinity outperformed structure-based models on the S34 benchmark test set, which is a meaningful result given that S34 specifically tests generalization to novel peptide-protein pairs.
The tradeoff is interpretability. Sequence models tell you what the predicted affinity is, but they offer limited mechanistic insight into which residues drive binding. For lead optimization where you need to know exactly where to mutate, that limitation matters.
Structure-based approaches and docking
Structure-based methods use 3D coordinates to model peptide-protein interactions physically. Peptide docking algorithms like AutoDock CrankPep, HADDOCK, and Rosetta FlexPepDock sample conformational space and score poses using energy functions. The problem is that conventional docking scoring functions are notoriously inaccurate for absolute affinity prediction, with Pearson correlation coefficients around 0.316 for peptide systems.

MM/PBSA and MM/GBSA post-processing rescoring changes that picture substantially. Optimized MM/PBSA(GBSA) methods achieve Rp values of 0.732, more than double the predictive performance of raw docking scores. The computational cost is higher, roughly 3 seconds per complex for optimized implementations, but that is entirely tractable for focused lead sets.
Hybrid and deep learning frameworks
The most capable current systems integrate sequence context, structural geometry, and graph-based representations into unified architectures. ConGA-PepPI achieved an accuracy of 0.839 and AUROC of 0.921 with approximately 3 seconds per prediction, which places it in a genuinely useful operational range for drug discovery pipelines. The model combines graph neural networks with attention-based sequence encoding to capture both local residue contacts and global interaction topology.
Other frameworks like CMHS incorporate multimodal learning, fusing sequence, structural, and physicochemical features. B-factor-guided noise has been incorporated into graph-based models to better represent binding-induced flexibility, which is one of the most persistent failure modes in static structure-based prediction.
| Method class | Accuracy | Speed | Data requirement |
|---|---|---|---|
| Sequence-based (e.g., CrossAffinity) | Moderate to high | Very fast | Sequence only |
| Docking + MM/PBSA | High (with refinement) | Moderate | 3D structure required |
| Hybrid deep learning (e.g., ConGA-PepPI) | High | Fast | Sequence + structure |
Pro Tip: For targets where AlphaFold3 structures are your only structural input, validate the predicted binding interface against known mutagenesis data before using it as a docking template. AlphaFold3 encodes interaction-aware geometries, but static predicted structures can still mislead affinity scoring for flexible binding sites.
Building a tiered prediction workflow
The single biggest practical improvement you can make to your affinity prediction pipeline is switching from a single-method approach to a layered workflow. Experts consistently recommend pairing ML speed with physics-based rigor for realistic, scalable drug discovery.
Here is how to structure that workflow:
-
Initial ML screening. Run your full candidate library through a sequence-based or hybrid deep learning model. At this stage, you are filtering, not ranking precisely. Set a permissive affinity threshold and carry forward the top 10 to 20% of candidates.
-
Structural preparation. For the filtered set, generate or retrieve 3D structures. Use AlphaFold3 for novel targets, or retrieve PDB structures when available. Prepare structures with protonation state assignment at physiological pH and energy minimization before docking.
-
Peptide docking. Apply a peptide docking algorithm appropriate to your target class. For flexible peptides above 10 residues, ensemble docking with multiple starting conformations reduces pose-sampling bias.
-
MM/PBSA or FEP refinement. Rescore the top docked poses using MM/PBSA(GBSA) or, for highest-priority candidates, free energy perturbation (FEP). This step is where you get the quantitative affinity estimates reliable enough to inform synthesis decisions.
-
Uncertainty quantification. Apply Bayesian uncertainty estimation to flag predictions where the model is operating outside its training distribution. Bayes by Backprop provides superior calibration for deep learning models without requiring separate recalibration steps.
-
Prioritization and experimental handoff. Rank candidates by predicted affinity combined with uncertainty score. High confidence, high affinity predictions go to synthesis first. Flag high uncertainty predictions for additional structural analysis before committing resources.
The most common pitfall at this stage is treating ML scores and MM/PBSA scores as directly comparable. They are not. ML scores are relative rankings within a training distribution. MM/PBSA scores approximate physical free energies. Mixing them without normalization produces incoherent rankings. Keep them in separate decision layers until you have calibrated their relationship on a validated internal dataset.
Interpreting results and validation strategies
A predicted affinity value without a confidence estimate is a number you cannot act on responsibly. Here is what to look at when evaluating your outputs:
- Pearson correlation coefficient (Rp) and Spearman rank correlation (Rs): These measure how well predicted values track experimental measurements. An Rp above 0.7 on an external test set is considered strong for peptide systems.
- Root mean square error (RMSE): Expressed in kcal/mol for free energy predictions. RMSE below 1.5 kcal/mol is generally acceptable for lead prioritization.
- AUROC for binary classification tasks: When the goal is to separate binders from non-binders rather than rank affinities precisely, AUROC above 0.85 indicates a reliable classifier.
- Uncertainty intervals: Predictions with wide Bayesian credible intervals signal that the model is extrapolating. Uncertainty quantification is often overlooked but is critical for avoiding false positives in virtual screening campaigns.
- Cross-validation strategy: Use stratified k-fold cross-validation with sequence identity clustering to avoid data leakage. External benchmarking on held-out datasets like S34 provides the most honest performance estimate.
Experimental validation remains the final arbiter. Surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and fluorescence polarization assays each provide Kd values that you can directly compare against predicted affinities. Build a feedback loop where experimental results update your model's training data. Even a small set of 20 to 50 validated measurements on your specific target class can substantially improve prediction accuracy for that system.
My perspective on where affinity prediction actually breaks down
I have seen a consistent pattern in how teams approach predicting peptide interactions: they pick one method, optimize it heavily, and then express genuine surprise when it fails on a novel target class. In my experience, the failure is almost never the algorithm. It is the assumption that a model trained on diverse benchmark data will generalize to your specific, often unusual, protein family.
The cold start problem is real. When you are working with a protein target that has fewer than 10 known peptide binders in any public database, sequence-based models are essentially guessing. Physics-based methods are your only credible option, and even those require careful structural preparation. I have found that combining sequence-based screening with MM/PBSA refinement, even when the sequence model's absolute predictions are noisy, still outperforms either method alone because the errors are partially uncorrelated.
What I think is genuinely underappreciated is the role of conformational dynamics. Static structures, including AlphaFold3 outputs, represent one snapshot of a protein that exists as an ensemble of conformations in solution. Binding-induced conformational changes can shift affinity by orders of magnitude, and models that ignore this will systematically misrank flexible binding sites. Incorporating B-factor information or running short MD simulations before docking is not optional for these targets. It is the difference between a useful prediction and a misleading one.
My advice for researchers starting out: do not chase the highest-accuracy model on a leaderboard. Chase the model that fails predictably. A method with known failure modes you can work around is worth far more than a black box with impressive benchmark numbers.
— Hooman
How Innovabiotech supports your prediction workflows
Innovabiotech's team specializes in exactly the kind of multi-method, tiered workflows described in this guide. Whether you need custom peptide design with integrated affinity prediction, or a full virtual screening pipeline combining ML scoring with physics-based refinement, Innovabiotech builds workflows calibrated to your specific target and data context.

From de novo peptide design through hit-to-lead optimization and experimental validation support, Innovabiotech's computational biology team works directly with your project data. Every workflow is built around your target class, not a generic template. If you are ready to move from benchmark performance to real-world prediction accuracy, reach out to Innovabiotech for a project consultation.
FAQ
What is peptide binding affinity prediction?
Peptide binding affinity prediction uses computational methods to estimate how strongly a peptide binds to a target protein, expressed as a dissociation constant (Kd) or free energy of binding. Methods range from sequence-based machine learning models to physics-based MM/PBSA calculations.
When should I use sequence-based vs. structure-based methods?
Use sequence-based models when high-resolution structural data is unavailable or when screening large libraries at speed. Structure-based methods like docking with MM/PBSA refinement are better suited for lead optimization where quantitative affinity estimates and binding pose information are required.
How accurate are current affinity prediction methods?
Optimized MM/PBSA methods achieve Pearson correlations around 0.73 for peptide systems, while hybrid deep learning frameworks like ConGA-PepPI reach AUROC values of 0.921. Accuracy depends heavily on data quality and whether the target falls within the model's training distribution.
Why is uncertainty quantification important in binding affinity modeling?
Uncertainty estimates identify predictions where the model is extrapolating beyond its training data. Bayesian methods like Bayes by Backprop provide calibrated confidence intervals that help you avoid acting on false positives, which is particularly critical in high-throughput virtual screening campaigns.
How do I validate computational affinity predictions experimentally?
Surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) are the standard experimental methods for measuring Kd values that can be directly compared against computational predictions. Building a feedback loop between experimental results and model retraining improves accuracy for your specific target class over time.
Recommended
- Innova Biotech Solutions: Expert Peptide Design and Optimization Services.bioinformatics validation, custom peptides, peptide design services
- Virtual Screening
- Protein engineering, Chimeric Protein Design Services, computational modeling
- Biotechnology Advancements: Innovations at Innova Biotech Solutions