
Peptide secondary structure is defined as the localized, regularly repeating folding of the polypeptide backbone into alpha-helices, beta-sheets, and related motifs, stabilized by backbone hydrogen bonds between amide N–H donors and carbonyl C=O acceptors. These structural elements sit above primary sequence in the protein hierarchy but below tertiary and quaternary organization, making them the critical intermediate layer where sequence information translates into three-dimensional form. Understanding what defines secondary structure at the atomic level is not optional background knowledge for structural biologists. It is the foundation on which protein engineering, drug design, and de novo peptide design are built. This article covers the molecular chemistry, motif types, assignment methods, and functional significance of peptide secondary structure with the precision that bench and computational researchers require.
What is peptide secondary structure at the molecular level?
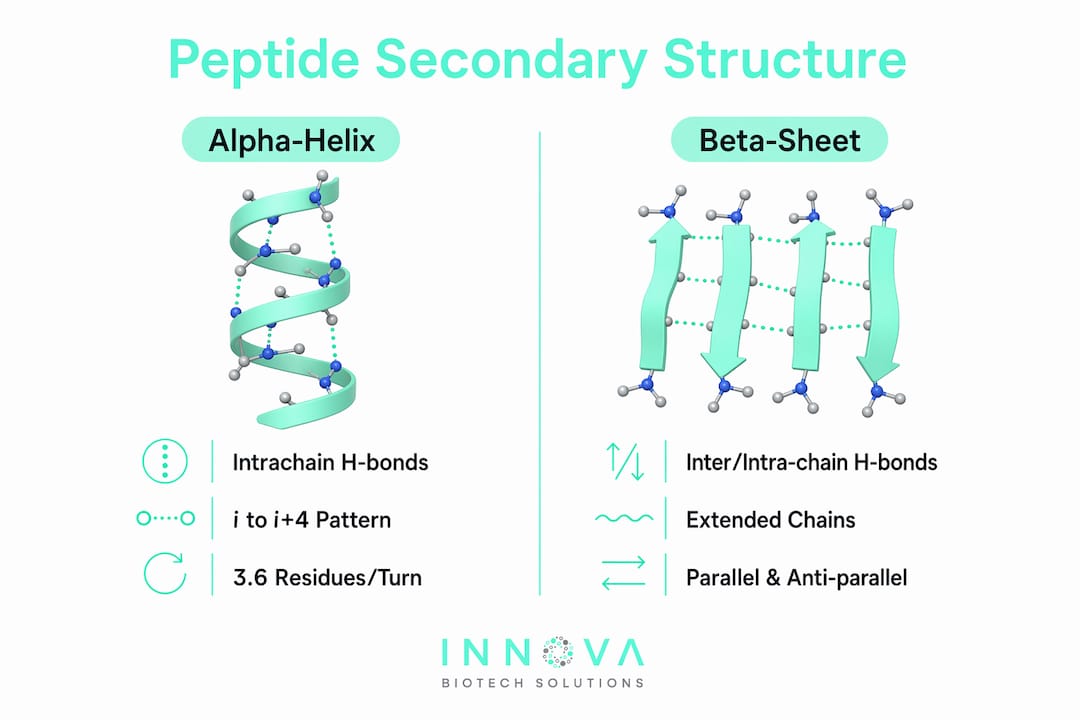
Secondary structure is defined by the geometry of backbone hydrogen bonding, not by side-chain chemistry. The polypeptide backbone carries repeating amide N–H groups and carbonyl C=O groups at every residue. When these groups form hydrogen bonds in a regular, repeating pattern along the chain, the backbone adopts a stable, predictable conformation. Side-chain conformations are explicitly excluded from the formal definition of secondary structure, even though they influence which conformations are energetically preferred.
The four levels of protein structure are primary (amino acid sequence), secondary (local backbone folding), tertiary (overall three-dimensional fold), and quaternary (multi-chain assembly). Secondary structure occupies the second tier because it emerges directly from local sequence propensities and backbone geometry, before long-range interactions between distant residues shape the tertiary fold. This hierarchy matters practically: you can disrupt tertiary structure with a single buried mutation while leaving secondary elements largely intact, because tertiary structure is more sensitive to mutations than secondary structure is.

The two dominant secondary structure types are the alpha-helix and the beta-sheet. Both arise from the same chemical logic: the backbone amide and carbonyl groups satisfy their hydrogen-bonding capacity through intrachain or interchain contacts, reducing the energetic cost of burying the backbone in a hydrophobic core. Loops, turns, and coil regions complete the picture, connecting helices and sheets without forming regular hydrogen-bond patterns.
Why backbone hydrogen bonds define the structure
Individual backbone hydrogen bonds are modest in strength, typically in the range of 2 to 10 kcal/mol. Their power comes from repetition. The repeated hydrogen-bonding network in alpha-helices and beta-sheets creates a collectively strong stabilizing force that no single bond could achieve alone. This geometric cooperativity is why disrupting one hydrogen bond in a helix rarely unfolds the entire element, but disrupting several in sequence can.
Pro Tip: When interpreting molecular dynamics trajectories, track the fraction of backbone hydrogen bonds satisfied over time rather than relying on instantaneous snapshots. A helix that appears intact in a single frame may be transiently disrupted across the ensemble.
What are the main types of peptide secondary structure?
The alpha-helix and the beta-sheet account for the majority of regular secondary structure in folded proteins. Each has distinct geometric characteristics that determine how it packs, how it interacts with ligands, and how it responds to sequence changes.
Alpha-helix geometry and features
The alpha-helix is stabilized by an i to i+4 hydrogen-bond pattern, where the carbonyl oxygen of residue i bonds to the amide hydrogen of residue i+4. Crystal structure evidence confirms 3.6 residues per turn in a right-handed coil, with a helical rise of approximately 1.5 Å per residue. The side chains project outward from the helical axis, leaving the backbone hydrogen-bond network fully internal. This geometry makes alpha-helices compact, regular, and highly amenable to computational prediction.
Deviations from canonical geometry matter. Peptide bond planarity and protonation state changes can distort or weaken helices in both experimental structures and simulations. A helix that looks intact by visual inspection may carry strained bond angles that DSSP-style algorithms flag as non-helical. This is one reason visual assignment and algorithmic assignment do not always agree.
Beta-sheet geometry and features
Beta-sheets are stabilized by hydrogen bonds between aligned beta strands running either parallel or antiparallel. In antiparallel sheets, the hydrogen bonds are more linear and therefore geometrically stronger. In parallel sheets, the bonds are slightly bent, which reduces individual bond strength but allows for larger, more extended sheet structures. Beta-sheets are the dominant structural element in immunoglobulins, beta-barrel membrane proteins, and amyloid fibrils.
Beyond helices and sheets, several additional motifs complete the peptide conformation catalog:
- Beta-turns: Four-residue motifs that reverse chain direction, often connecting antiparallel beta strands. They are classified into Types I, II, I', and II' based on the phi/psi angles of the central two residues.
- Beta-hairpins: The simplest supersecondary motif, consisting of two antiparallel beta strands connected by a short loop or turn.
- Loops and coils: Irregular regions that lack repeating hydrogen-bond patterns. They are not random; they adopt preferred conformations and are often functionally critical at active sites and binding interfaces.
Pro Tip: Do not treat loop regions as structurally unimportant. Many enzyme active sites and antibody complementarity-determining regions (CDRs) are loops. Their conformational flexibility is a feature, not a deficiency.
The table below compares the key structural features of alpha-helices and beta-sheets:
| Feature | Alpha-helix | Beta-sheet |
|---|---|---|
| H-bond pattern | Intrachain, i to i+4 | Interchain or intrastrand |
| Strand/residue geometry | 3.6 residues/turn, right-handed | Parallel or antiparallel strands |
| Side-chain orientation | Radially outward | Alternating above/below sheet plane |
| Common occurrence | Membrane proteins, coiled coils | Immunoglobulins, beta-barrels, amyloids |
| Sensitivity to proline | Helix-breaking residue | Tolerated at strand edges |
How is secondary structure assigned and analyzed?
Secondary structure assignment is not a visual exercise. The gold standard is DSSP-style algorithmic assignment, which classifies backbone hydrogen bonds using precise geometric criteria derived from atomic-resolution coordinates. DSSP (Dictionary of Secondary Structure of Proteins) defines a hydrogen bond as present when the electrostatic interaction energy between the N–H and C=O groups falls below a defined threshold, accounting for bond distance and angle. This approach removes subjectivity and produces reproducible assignments across different research groups.
The primary experimental sources for atomic-resolution coordinates are:
- X-ray crystallography: Provides high-resolution electron density maps from which backbone coordinates are derived. Resolution below 2.0 Å is generally required for reliable hydrogen-bond geometry assignment.
- NMR spectroscopy: Yields solution-state structural ensembles. Secondary structure is inferred from chemical shift indices (CSI), NOE patterns, and coupling constants. NMR captures conformational dynamics that crystal structures cannot.
- Cryo-electron microscopy (cryo-EM): Increasingly capable of near-atomic resolution for large complexes. Secondary structure elements are identifiable at resolutions of 3 to 4 Å, though precise hydrogen-bond geometry requires higher resolution.
Computational prediction methods, including tools such as PSIPRED, JPred4, and ESMFold, assign secondary structure from sequence alone using evolutionary information and deep learning. These tools are valuable for rapid screening but carry error rates that make experimental validation necessary for high-confidence structural claims. Accurate secondary structure assignments rely on precise hydrogen bonding patterns rather than backbone dihedral angles or visual inspection alone.
Why does secondary structure matter for folding, function, and stability?
Secondary structure elements are the building blocks of tertiary folds, and they form early. Local ordering such as beta-hairpins precedes complete tertiary folding, meaning secondary structure acts as a scaffold that nucleates the final three-dimensional architecture. This early formation has direct implications for protein folding diseases: misfolded beta-sheet aggregates in amyloid fibrils, as seen in Alzheimer's disease and Parkinson's disease, arise when secondary structure elements assemble incorrectly before the tertiary fold can suppress aggregation-prone conformations.
The functional consequences of secondary structure extend across protein classes:
- Structural proteins: Alpha-keratin in hair and nails is built from coiled-coil alpha-helices. Silk fibroin is dominated by antiparallel beta-sheets. The mechanical properties of these materials arise directly from secondary structure geometry.
- Enzymes: The TIM barrel fold, one of the most common enzyme architectures, consists of eight alternating alpha-helices and beta-strands. The active site geometry depends on precise positioning of these elements.
- Membrane proteins: Alpha-helical bundles span lipid bilayers in G-protein-coupled receptors (GPCRs) and ion channels. The hydrophobic periodicity of the helix surface is what drives membrane insertion.
- Antibodies: The immunoglobulin fold is a beta-sandwich, and the CDR loops that determine antigen specificity are positioned by the underlying sheet framework.
Secondary structure elements also contribute to protein stability through their hydrogen-bonding networks. A helix buried in a hydrophobic core satisfies backbone hydrogen-bond capacity internally, avoiding the energetic penalty of exposing unsatisfied polar groups to a nonpolar environment. This thermodynamic logic explains why helices are overrepresented in membrane-spanning regions and why beta-sheets are common in soluble proteins where strand-strand contacts can form across the aqueous interface.
For researchers working on protein stability engineering or therapeutic peptide design, secondary structure propensity is a primary design variable. Substituting a helix-breaking proline into a critical alpha-helix, or introducing a beta-sheet-favoring valine into a loop, can shift the conformational equilibrium measurably. Side-chain chemistry strongly biases the propensity for regions to form alpha-helices or beta-sheets, even though side chains are formally excluded from the secondary structure definition.
Key takeaways
Peptide secondary structure is the foundational layer of protein architecture, defined by backbone hydrogen-bond geometry and expressed as alpha-helices, beta-sheets, and connecting motifs that collectively determine folding, stability, and function.
| Point | Details |
|---|---|
| Definition is backbone-specific | Secondary structure is defined by N–H to C=O hydrogen bonds in the backbone, not side-chain interactions. |
| Two dominant motifs | Alpha-helices (i to i+4 bonding, 3.6 residues/turn) and beta-sheets (parallel or antiparallel strands) account for most regular secondary structure. |
| Assignment requires geometry | DSSP-style algorithms using precise H-bond geometry outperform visual or dihedral-angle-only methods for reliable assignment. |
| Secondary structure forms first | Local secondary elements like beta-hairpins nucleate early in folding, scaffolding the final tertiary architecture. |
| Side chains influence but do not define | Side-chain chemistry biases secondary structure propensity without being part of the formal definition. |
The part most researchers underestimate
Most researchers I work with understand alpha-helices and beta-sheets at the level of a textbook diagram. Where the real complexity lives is in the gap between a static secondary structure assignment and the dynamic conformational ensemble that actually governs function. A DSSP assignment from a single crystal structure tells you what the backbone was doing in one crystal packing environment at one temperature. It does not tell you what that region does in solution, under physiological salt conditions, or when a ligand binds.
The second misconception I encounter regularly is treating secondary structure as a fixed property of a sequence. Peptide conformation is context-dependent. The same 10-residue sequence can adopt a helix in one protein environment and a strand in another, depending on neighboring residues, solvent exposure, and binding partners. This is not a failure of prediction algorithms. It is a real physical phenomenon, and it means that secondary structure propensity scores from tools like PSIPRED are probabilities, not certainties.
My practical recommendation: always pair computational secondary structure prediction with at least one experimental validation, whether that is circular dichroism (CD) spectroscopy for bulk helical content, NMR chemical shift indexing for residue-level assignment, or cryo-EM for large assemblies. The peptide binding affinity and structural behavior you observe experimentally will often surprise you relative to the predicted conformation, and that surprise is where the most interesting biology lives.
— Hooman
How Innovabiotech supports your peptide structure research
Translating secondary structure knowledge into functional peptide design requires more than sequence intuition. It requires validated computational modeling, experimental feedback loops, and domain expertise in backbone geometry and folding dynamics.

Innovabiotech provides custom peptide design services that integrate secondary structure analysis, bioinformatics validation, and de novo design workflows tailored to your research objectives. Whether you are optimizing a therapeutic peptide for helical stability, designing a beta-sheet scaffold for a binding interface, or engineering a novel fold from scratch, the Innovabiotech team brings the structural biology depth your project requires. For researchers who also need protein engineering support at the tertiary level, Innovabiotech's computational modeling services cover the full structural hierarchy. Contact Innovabiotech to discuss how secondary structure-informed design can accelerate your research outcomes.
FAQ
What is peptide secondary structure in simple terms?
Peptide secondary structure is the local, regular folding of a polypeptide backbone into alpha-helices, beta-sheets, and related motifs, stabilized by hydrogen bonds between backbone amide N–H and carbonyl C=O groups. Side-chain conformations are not part of this definition.
What are the two main types of secondary structure in proteins?
The two primary types are the alpha-helix, stabilized by i to i+4 intrachain hydrogen bonds with 3.6 residues per turn, and the beta-sheet, stabilized by hydrogen bonds between parallel or antiparallel backbone strands.
How is secondary structure different from tertiary structure?
Secondary structure describes local, regular backbone folding patterns within a single region of the chain. Tertiary structure describes the overall three-dimensional arrangement of all secondary elements and side chains across the entire polypeptide, and it is more sensitive to mutations than secondary structure is.
How do researchers assign secondary structure experimentally?
X-ray crystallography, NMR spectroscopy, and cryo-EM provide atomic coordinates from which DSSP-style algorithms assign secondary structure based on precise backbone hydrogen-bond geometry, which is more reliable than visual inspection or dihedral angle criteria alone.
Why does secondary structure matter for drug and peptide design?
Secondary structure elements form early in folding and act as scaffolds for tertiary architecture, meaning that engineering or disrupting specific helices or sheets directly controls protein stability, binding geometry, and function. For researchers sourcing research peptides for structural studies, understanding the target conformation is prerequisite to selecting or designing the right sequence.
Recommended
- Innova Biotech Solutions: Expert Peptide Design and Optimization Services.bioinformatics validation, custom peptides, peptide design services
- Peptide Binding Affinity Prediction: A Practical Guide
- Protein engineering, Chimeric Protein Design Services, computational modeling
- Why Protein Stability Engineering Is Critical for Drug Development