
Bioinformatics peptide optimization is the computational tailoring of peptide sequences to improve therapeutic properties including binding affinity, stability, solubility, and pharmacokinetics before synthesis. Known formally as computational peptide design, this discipline combines machine learning, structural bioinformatics, and molecular simulation to predict sequence-to-function relationships at a scale no wet-lab workflow can match. Platforms like PepAnno, CreoPep, and PepTune now represent the operational standard for teams running drug discovery pipelines. The role of bioinformatics in peptide optimization has shifted from supporting tool to primary driver of candidate selection, compressing timelines and raising hit rates across therapeutic programs.
What computational methods are foundational for peptide optimization?
The core model architectures driving peptide optimization span both sequence-based and structure-based representations, each capturing different layers of biological information. Selecting the right architecture depends on the data available and the therapeutic objective.
Sequence-based models dominate early-stage filtering:
- Convolutional neural networks (CNNs) detect local sequence motifs and residue patterns associated with activity or toxicity.
- BiLSTM and BiGRU networks capture long-range dependencies across the peptide chain, critical for understanding how distant residues influence folding and function.
- Attention mechanisms assign importance weights to individual residues, making model decisions interpretable for medicinal chemists.
- Graph neural networks (GNNs) operate on contact maps derived from 3D structural data, encoding spatial relationships that sequence alone cannot represent.
Traditional ML retains value for interpretability, while deep learning captures complex nonlinear relationships in sequence and structure. Modern peptide optimization frameworks blend both for robustness, a hybrid approach that outperforms either method in isolation.
Generative models represent the frontier of bioinformatics in peptide design. CreoPep, developed for conotoxin optimization, uses a conditional generative masked-language model combined with temperature-controlled multinomial sampling and iterative physics-based binding energy screening. This architecture generates novel sequences with target-specific potency rather than simply scoring existing ones. PepTune extends this logic to multi-objective generation, applying Monte Carlo Tree Search-guided masked discrete diffusion to produce Pareto non-dominated sequences across binding affinity, solubility, hemolysis, and membrane permeability simultaneously.
Pro Tip: When selecting a model architecture, match the representation to your data type. If you have solved structures, GNNs on contact maps will outperform sequence-only models. If structural data is sparse, attention-based sequence models with transfer learning from large protein language models are the practical starting point.
How do integrated multi-stage pipelines optimize peptides for therapeutic profiles?
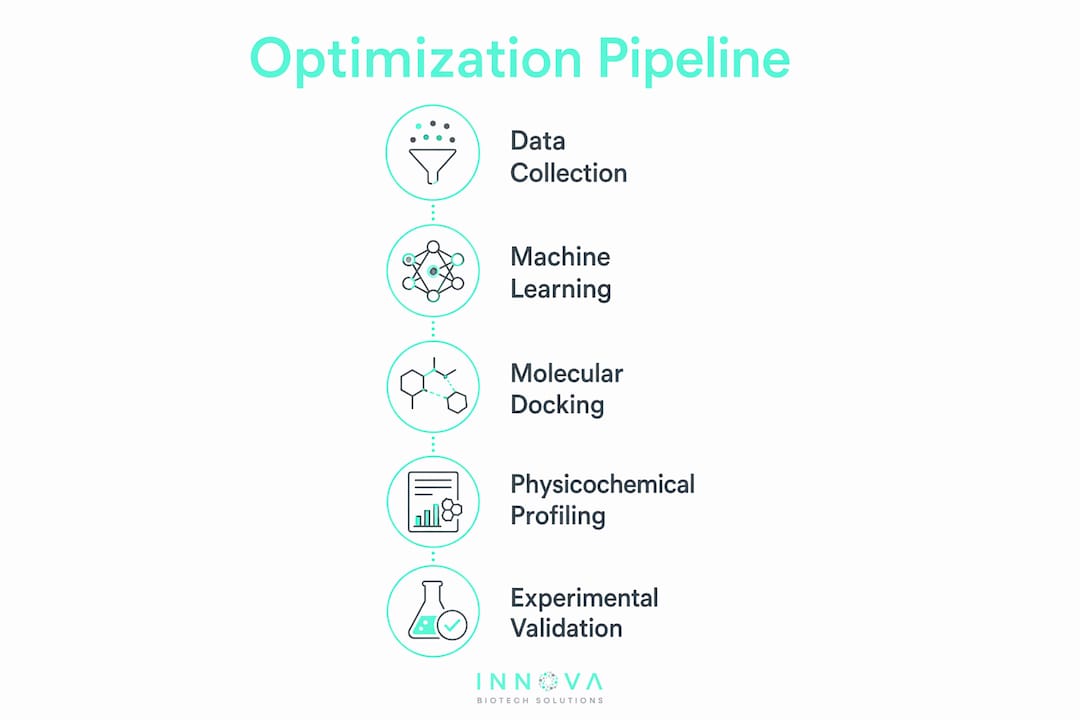
A single model rarely delivers a clinical candidate. The real power of computational peptide sequence optimization comes from chaining multiple analytical stages into a coherent pipeline that filters candidates against all relevant therapeutic criteria simultaneously.
A well-constructed end-to-end pipeline typically follows this sequence:
- Archive mining and data curation. Structured databases are queried for known active sequences, physicochemical profiles, and experimental assay results. Data quality at this stage determines model performance downstream.
- ML predictive scoring. Candidate sequences are scored for potency, hemolysis, stability, and other properties using trained classifiers or regressors. This step eliminates the majority of unqualified sequences before any structural analysis.
- Generative AI sequence design. Models like PepTune or CreoPep propose novel sequences that satisfy the scoring thresholds established in step two, expanding the search space beyond known analogs.
- Molecular docking. Top candidates are docked against the target protein to assess binding pose and interaction geometry, providing a mechanistic filter that sequence models cannot replicate.
- Molecular dynamics (MD) simulation. Shortlisted candidates undergo MD to evaluate conformational stability, membrane interaction, and binding persistence under physiological conditions.
- Experimental validation. Electrophysiological assays, surface plasmon resonance, or cell-based assays confirm computational predictions before advancing to lead optimization.
The synergy between ML and docking/MD is not optional. Generative AI alone produces sequences that score well computationally but fail biologically when conformation and membrane context are ignored. Multiscale modeling acts as the mechanistic reality check that keeps the pipeline grounded.
Pro Tip: Do not treat molecular docking as a final validation step. Run docking early as a secondary filter after ML scoring to eliminate structurally implausible candidates before committing to the computational cost of full MD simulations.

PepAnno addresses a different bottleneck. Its one-click automated pipeline integrates physicochemical profiling, structure prediction, and functional annotation in a single workflow, reducing the manual handoffs between analysis stages that fragment most research programs. Workflow fragmentation limits project throughput more than model accuracy in most operational settings.
What are the practical applications of peptide optimization in drug discovery?
The impact of bioinformatics on peptides is measurable in experimental outcomes, not just theoretical capability. Several documented cases illustrate what these technologies deliver in practice.
The most striking result comes from venom peptide research. A machine learning and high-throughput platform achieved a 100% hit rate in binder discovery across four distinct molecular targets. This is not a marginal improvement over traditional screening. It represents a categorical shift in how early-stage discovery operates when ML models replace random library screening.
Affinity maturation timelines have also been compressed significantly. ML-assisted affinity maturation improved binding affinity up to 500-fold within a single evolutionary cycle using a supervised learner trained on enrichment data. Traditional directed evolution requires multiple iterative rounds to achieve comparable gains, each round adding weeks of experimental time.
Computational peptide optimization does not replace experimental biology. It reorders the workflow so that experiments confirm predictions rather than generate them, concentrating wet-lab resources on candidates that have already passed rigorous computational filters.
Additional application areas where these methods deliver documented value include:
- Antimicrobial peptide (AMP) design, where ML models trained on membrane-disruption data generate sequences with activity against drug-resistant pathogens.
- Conotoxin optimization via CreoPep, targeting ion channels and receptors with high selectivity requirements.
- Cell-penetrating peptide (CPP) engineering, where permeability and cargo delivery efficiency are co-optimized alongside cytotoxicity profiles.
- Developability assessment using platforms like PeptiVerse, which evaluates drug-like properties for canonical sequences and modified peptide SMILES early in the program to reduce late-stage attrition.
Integration with protein engineering workflows extends these gains further. When peptide optimization is coupled with structural protein modeling, teams can design binders that exploit specific allosteric sites or interface geometries identified through computational protein analysis, a capability that purely sequence-based approaches cannot access.
What are the key challenges and best practices in bioinformatics-driven workflows?
Bioinformatics peptide optimization carries well-documented failure modes that experienced practitioners plan around from the start.
The most common problems are:
- Data imbalance. Most peptide datasets contain far more inactive sequences than active ones, biasing classifiers toward false negatives on novel scaffolds.
- Conformation blindness. Sequence-only models miss activity dependencies that arise from secondary structure or membrane-bound conformations.
- Single-metric gaming. Optimizing for binding affinity alone produces candidates with poor solubility, hemolysis risk, or metabolic instability.
- Workflow fragmentation. Disconnected tools for scoring, docking, and annotation create manual handoffs that slow iteration and introduce transcription errors.
Pareto optimization addresses the single-metric problem directly. PepTune generates Pareto non-dominated sequences, meaning no candidate in the output set can be improved on one objective without degrading another. This formalization prevents the common failure mode where a peptide scores perfectly on affinity but fails every other developability criterion.
For data quality, physics-based pseudo-labeling during iterative data augmentation improves effective training signals more reliably than architecture changes alone. CreoPep's approach of aligning training data with binding energy calculations from physics-based screening connects the model directly to mechanism, not just statistical correlation.
Pro Tip: Before scaling your pipeline, audit your training labels. Mislabeled or loosely defined activity thresholds are the single largest source of model underperformance in peptide ML projects. Tightening label definitions around a specific assay and concentration cutoff consistently outperforms switching to a more complex architecture.
Experimental validation remains non-negotiable. Computational scores predict biological behavior; they do not confirm it. Electrophysiological assays, as used in CreoPep validation, and cell-based permeability assays for CPPs provide the ground truth that keeps computational models calibrated over successive project iterations. You can explore how peptide library screening integrates with these computational workflows to further sharpen candidate selection.
Key takeaways
Bioinformatics peptide optimization works because hybrid AI architectures, multi-stage pipelines, and Pareto-based multi-objective scoring together convert sequence data into experimentally validated therapeutic candidates faster than any single-method approach.
| Point | Details |
|---|---|
| Hybrid model architecture | Combining CNN, BiLSTM, attention, and GNN captures both sequence and structural information for stronger predictions. |
| Multi-stage pipeline design | Chaining ML scoring, generative design, docking, and MD simulation filters candidates against all therapeutic criteria before synthesis. |
| Pareto multi-objective optimization | PepTune-style Pareto generation prevents single-metric gaming across affinity, solubility, hemolysis, and permeability. |
| Physics-based data augmentation | Pseudo-labeling with binding energy calculations improves training quality more reliably than architectural complexity alone. |
| Experimental validation is mandatory | Computational scores must be confirmed by assays; the pipeline reorders experiments, it does not eliminate them. |
Why the field still underestimates integrated workflows
Most discussions of computational peptide design focus on model architecture. After working across multiple drug discovery programs, I find that architecture is rarely the limiting variable. The bottleneck is almost always workflow integration.
Teams running disconnected tools for scoring, docking, annotation, and reporting spend more time on data transfer and format conversion than on scientific decision-making. PepAnno's one-click pipeline is not impressive because of its underlying model. It is impressive because it removes the friction that kills project momentum between analysis stages. That operational insight is worth more than any marginal accuracy gain from a newer transformer variant.
The shift from trial-and-error peptide synthesis to data-driven candidate prioritization is real and documented. A 100% hit rate in binder discovery and 500-fold affinity improvement in a single evolutionary cycle are not theoretical projections. They are published outcomes from teams that committed to full pipeline integration rather than point-solution adoption.
My recommendation for any pharma or biotech team entering a peptide program in 2026: invest in de novo peptide design frameworks that connect generative models directly to mechanistic validation, and treat multi-objective Pareto optimization as the default formulation, not an advanced option. The teams that will compress timelines most aggressively are those that stop optimizing individual tools and start optimizing the connections between them.
— Hooman
Accelerate your peptide programs with Innovabiotech

Innovabiotech, based in San Francisco, provides end-to-end computational peptide design and optimization services built for pharmaceutical and biotech drug discovery teams. From ML-based sequence scoring and generative peptide design to molecular docking, MD simulation, and multi-objective candidate prioritization, the team delivers integrated pipelines tailored to your therapeutic target and program stage. Whether you are working on antimicrobial peptides, conotoxins, cell-penetrating peptides, or receptor-targeted binders, Innovabiotech's peptide design services combine bioinformatics rigor with wet-lab-ready outputs. The team also offers protein engineering and computational modeling to support programs where peptide optimization intersects with structural protein design.
FAQ
What is the role of bioinformatics in peptide optimization?
Bioinformatics provides the computational methods, including ML models, structural prediction, and molecular simulation, that predict and improve peptide sequence-to-function relationships before synthesis. This reduces experimental cycles and raises hit rates in drug discovery programs.
What AI models are used in peptide sequence optimization?
CNNs, BiLSTM networks, attention mechanisms, and graph neural networks are the primary architectures, each suited to different data types and optimization objectives. Generative models like CreoPep and PepTune extend these capabilities to novel sequence design.
How does multi-objective optimization work for therapeutic peptides?
Platforms like PepTune use Pareto optimization to generate sequences that balance binding affinity, solubility, hemolysis, and membrane permeability simultaneously, preventing the common failure of optimizing one property at the expense of others.
How much can ML-assisted affinity maturation improve binding affinity?
ML-assisted affinity maturation has demonstrated up to 500-fold improvement in binding affinity within a single evolutionary cycle, compressing timelines that traditional directed evolution requires multiple rounds to achieve.
What is PepAnno used for in peptide research?
PepAnno is a structure-aware deep learning framework that automates physicochemical profiling, structure prediction, and functional annotation in a single pipeline, reducing manual workflow steps between analysis stages.
Recommended
- Innova Biotech Solutions: Expert Peptide Design and Optimization Services.bioinformatics validation, custom peptides, peptide design services
- Peptide Binding Affinity Prediction: A Practical Guide
- De Novo Peptide Design Benefits for Drug Discovery
- Examples of De Novo Protein Design: 2026 Research Guide