
TL;DR:
- Virtual screening uses computational methods to evaluate vast chemical libraries, accelerating drug discovery. It relies on structure-based or ligand-based approaches, enhanced by AI to improve speed and accuracy. Integrating virtual screening with experimental validation reduces costs and increases the likelihood of finding promising drug candidates.
Virtual screening services are defined as computational methods that evaluate millions to billions of chemical compounds against a biological target to identify the most promising drug candidates before any lab work begins. The industry-standard term is in-silico screening, and it sits at the front end of every modern drug discovery pipeline. Structure-Based Virtual Screening (SBVS) and Ligand-Based Virtual Screening (LBVS) are the two primary categories that define how these services operate. AI and machine learning have pushed screening speeds up to 10 million times faster than traditional docking methods, making computational compound screening the most cost-effective first filter in modern pharmaceutical research. Innovabiotech delivers these capabilities as tailored, project-specific services for biotech and pharmaceutical teams that need results they can act on.
What are virtual screening services and how do they work?
Virtual screening services use two distinct computational approaches, and choosing between them depends on what structural data you have available.

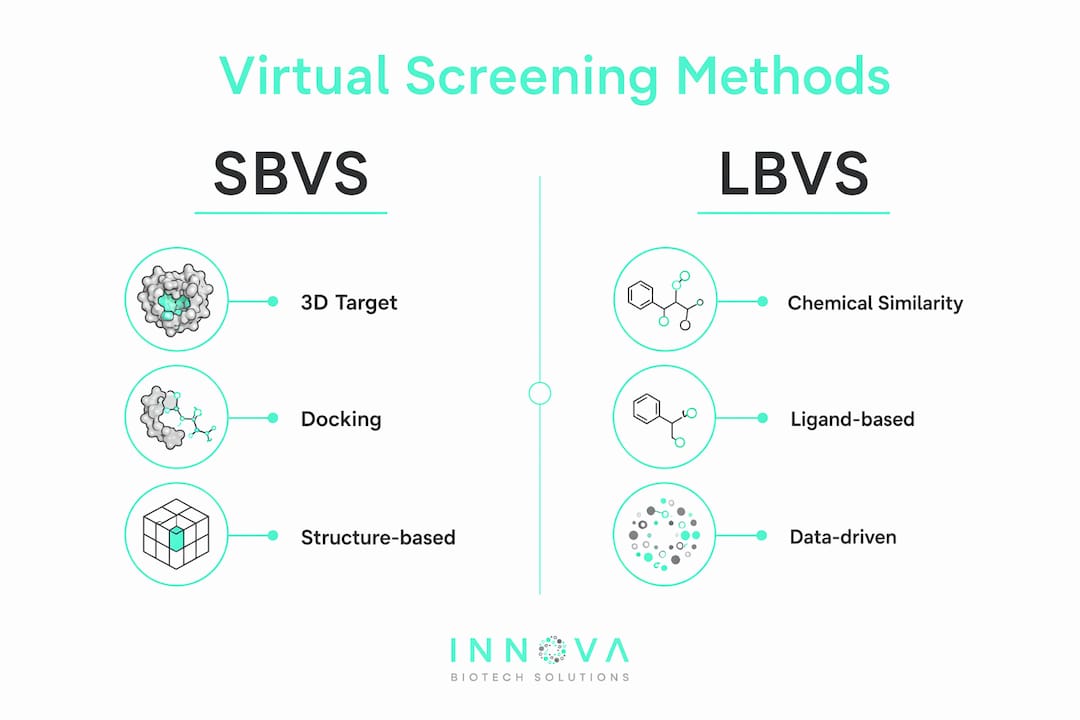
Structure-Based Virtual Screening (SBVS) requires a three-dimensional structure of the biological target, typically from X-ray crystallography or cryo-EM. The algorithm docks candidate compounds into the target's binding site and scores each pose based on predicted binding affinity. SBVS is the preferred method when high-quality target structures exist, because the spatial geometry of the binding pocket directly guides compound selection.
Ligand-Based Virtual Screening (LBVS) takes a different path. When no reliable target structure is available, LBVS uses the chemical and pharmacophoric properties of known active compounds to search for structurally or functionally similar candidates. Pharmacophore modeling and molecular fingerprint comparisons are the core tools here. LBVS is particularly useful for targets where structural data remains incomplete or ambiguous.
A standard virtual screening workflow follows four stages:
- Library preparation. Compounds are curated, filtered for basic physicochemical properties, and converted into 3D conformations ready for docking.
- Docking and scoring. Each compound is computationally placed into the target binding site and scored for predicted affinity and fit.
- Post-docking filtering. Hits are re-ranked using more computationally intensive methods such as molecular dynamics or free-energy calculations to reduce false positives.
- Compound prioritization. The top-ranked candidates are flagged for experimental validation, with the list sized to match the lab's throughput capacity.
Pro Tip: Balance computational cost against accuracy by running fast docking first to narrow a billion-compound library to a few thousand, then apply expensive free-energy calculations only to that shortlist. This staged approach cuts compute time without sacrificing hit quality.
AI and machine learning now sit inside every stage of this workflow. AI-driven compound prioritization improves both the speed and accuracy of post-screening data interpretation, which means fewer resources spent chasing dead ends.
What are the benefits of virtual screening in drug discovery?
The core benefit of computational compound screening is scale. Physical high-throughput screening (HTS) can test tens of thousands of compounds per day under ideal conditions. Virtual screening evaluates the same number in seconds and can process entire commercial libraries of hundreds of millions of compounds in days.
Cost reduction follows directly from that scale. Every compound that fails computationally is one fewer expensive assay run in the lab. Early triage through in-silico screening services concentrates wet-lab resources on candidates that already show computational promise, which lowers the per-hit cost of the entire discovery phase.
Key advantages biotech and pharmaceutical teams gain from virtual screening:
- Novel chemical space exploration. Computational methods can evaluate compounds that have never been synthesized, enabling scaffold hopping and the discovery of entirely new chemical series.
- Speed. AI frameworks achieve screening speeds up to 10 million times faster than conventional docking. That speed compresses timelines from years to weeks for the initial hit identification phase.
- Improved hit rates. Integrating virtual and physical screening iteratively raises the proportion of confirmed hits from the compounds that reach the lab.
- Reduced late-stage attrition. Filtering for ADMET properties computationally before synthesis prevents compounds with poor pharmacokinetics from consuming clinical resources.
The iterative integration of virtual and physical data is where the real efficiency gain appears. The most effective drug discovery strategy combines virtual screening with physical experimental screening in a data-driven feedback loop, not as two separate sequential steps.
What challenges do virtual screening services face?
Virtual screening is a hypothesis-generating process, not a definitive identification method. Virtual hits require iterative feedback loops with experimental data to confirm and refine predictions. Teams that treat computational hits as confirmed leads without lab validation consistently see high false-positive rates and wasted synthesis budgets.
The most common challenges in drug virtual screening include:
- False positives and false negatives. Scoring functions approximate binding affinity. They miss conformational flexibility, solvation effects, and protein dynamics that influence real binding behavior.
- Target model quality. An inaccurate or incomplete 3D structure produces misleading docking results. Homology models and low-resolution structures require additional validation before SBVS can be trusted.
- Library composition. A compound library that lacks chemical diversity will miss entire classes of potential hits, regardless of how good the docking algorithm is.
- Overly restrictive drug-likeness filters. Excessively restrictive filters can exclude novel but potentially potent compounds. Applying Lipinski's Rule of Five or similar heuristics too early in screening discards scaffolds that might succeed after optimization.
Advanced services address these limitations through multi-objective optimization, which considers potency, selectivity, and ADMET properties simultaneously rather than filtering for each criterion in isolation. This approach preserves more viable candidates through the early screening stages.
Pro Tip: Apply developability filters iteratively during lead optimization, not at the initial virtual screening stage. Early filtering for drug-likeness removes novel scaffolds before you know whether they can be optimized into viable leads. Save those filters for when you have experimental data to guide the decision.
Reducing false positives is a discipline in itself. Best practices for false positive reduction include ensemble docking across multiple target conformations, consensus scoring from two or more independent scoring functions, and mandatory experimental counter-screening for pan-assay interference compounds (PAINS).
How do virtual screening services integrate with AI and experimental workflows?
The most productive drug discovery pipelines treat computational and experimental screening as a single integrated system, not two separate departments. The cycle runs in three repeating phases: computational triage, experimental validation, and model update.
Modern virtual screening pipelines use integrated computational techniques including docking, pharmacophore modeling, molecular dynamics, and free-energy calculations to enhance enrichment and reduce synthetic burden. Each technique addresses a different weakness in the others. Docking is fast but approximate. Molecular dynamics captures protein flexibility that static docking misses. Free-energy calculations provide the most accurate binding predictions but require significant compute time.
The table below shows how different computational methods contribute to each phase of an integrated pipeline:
| Pipeline phase | Primary method | Key contribution |
|---|---|---|
| Initial triage | Fast docking | Reduces billion-compound libraries to thousands |
| Hit refinement | Pharmacophore modeling | Filters by 3D feature match, not just shape |
| Lead validation | Molecular dynamics | Captures binding stability over time |
| Affinity ranking | Free-energy calculations | Provides highest-accuracy binding predictions |
| ADMET prediction | Machine learning models | Flags absorption, distribution, metabolism, and toxicity risks early |
AI and machine learning sit across all five phases. AI-driven high-throughput screening enhances compound prioritization, image analysis, and post-screening data interpretation at every stage. Each round of experimental data feeds back into the predictive models, making the next computational cycle more accurate.
The practical benefit for biotech and pharmaceutical teams is a measurable reduction in the number of compounds that reach expensive late-stage assays without genuine potential. Validating virtual screening results through structured experimental feedback loops is the single most effective way to raise confidence in computational hits before committing synthesis resources.
Emerging methods are pushing accuracy further. Quantum chemistry calculations and cryo-EM guided ensemble docking represent the next frontier in virtual screening accuracy, particularly for targets with flexible binding sites or multiple relevant conformations. Teams that build these methods into their pipelines now will have a structural advantage as chemical libraries continue to grow in size and complexity.
The future of drug discovery depends on blending virtual screening's rapid hypothesis generation with biological validation to manage increasing chemical space and complexity. That blend is not optional. It is the only approach that scales.
Key Takeaways
Virtual screening services deliver the greatest value when computational triage, AI-driven model refinement, and experimental validation operate as a single integrated workflow rather than sequential steps.
| Point | Details |
|---|---|
| Two core methods | SBVS uses 3D target structures; LBVS uses chemical similarity when structural data is unavailable. |
| AI multiplies speed | AI frameworks screen compounds up to 10 million times faster than traditional docking, compressing hit identification timelines. |
| Hits are hypotheses | Virtual screening generates candidates for validation, not confirmed leads. Experimental feedback loops are required. |
| Filter timing matters | Apply drug-likeness filters during lead optimization, not at initial screening, to preserve novel scaffold diversity. |
| Integration is the strategy | Combining computational and physical screening iteratively raises hit rates and reduces late-stage attrition. |
What I've learned about virtual screening that most guides won't tell you
The biggest mistake I see biotech teams make is treating virtual screening as a black box that delivers answers. It does not. It delivers ranked hypotheses. The quality of those hypotheses depends entirely on the quality of the inputs: the target structure, the compound library, and the scoring model. Garbage in, garbage out is not a cliché here. It is a project-ending reality.
The second mistake is applying Lipinski filters at the start of a campaign and then wondering why the hits look like every other compound in the literature. Novel chemical space lives outside those filters. The teams that find genuinely differentiated scaffolds are the ones that screen broadly first and filter later, using experimental data to guide which heuristics actually matter for their specific target.
What actually works is building the feedback loop from day one. Run the computational screen, test a focused subset in the lab, feed those results back into the model, and run again. Each cycle narrows the search space and improves prediction accuracy. Three or four cycles of this process consistently outperform a single large virtual screen followed by a single large experimental screen.
The role of AI in this loop is real, but it is not magic. AI models are only as good as the training data behind them. If your experimental data is sparse or biased toward a narrow chemical series, the AI will reflect that bias. Data quality is the constraint that no algorithm can fix.
My recommendation for teams selecting in-silico screening services: ask the provider how they handle the feedback loop, not just how they run the initial screen. The answer tells you whether you are buying a one-time computation or a genuine discovery partnership.
— Hooman
Innovabiotech's computational drug discovery services
Biotech and pharmaceutical teams that need more than a one-time screen can work directly with Innovabiotech on fully integrated virtual screening programs built around their specific targets and compound libraries.

Innovabiotech combines structure-based and ligand-based screening with AI-driven model refinement, molecular dynamics, and ADMET prediction in a single coordinated workflow. The team also supports downstream hit-to-lead work through custom peptide design and protein engineering services, so the pipeline does not stop at the computational hit stage. Every project starts with a direct consultation to define the target, the library, and the validation strategy before any computation begins.
FAQ
What is the difference between SBVS and LBVS?
Structure-Based Virtual Screening uses a 3D model of the biological target to dock and score compounds, while Ligand-Based Virtual Screening uses the properties of known active compounds to find similar candidates when no reliable target structure exists.
How does virtual screening reduce drug discovery costs?
Virtual screening computationally filters millions of compounds before any lab assay runs, concentrating wet-lab resources on candidates that already show predicted activity and cutting the per-hit cost of the discovery phase.
Are virtual screening hits ready for synthesis?
Virtual screening hits are ranked hypotheses, not confirmed leads. Experimental validation through iterative feedback loops with wet-lab data is required before committing synthesis resources to any computational candidate.
How does AI improve virtual screening accuracy?
AI and machine learning models improve compound prioritization, scoring function accuracy, and post-screening data interpretation at every stage of the pipeline, with each round of experimental data making the next computational cycle more predictive.
What compound library size is needed for effective virtual screening?
Library size requirements depend on the target and the method, but modern in-silico screening services can evaluate libraries ranging from focused sets of tens of thousands to commercial collections exceeding one billion compounds, with AI-driven methods making the larger scales computationally practical.