
TL;DR:
- Virtual screening employs computational techniques to efficiently identify promising drug candidates from large chemical libraries. Combining structure-based and ligand-based methods enhances hit discovery, while multi-stage filtering reduces thousands of compounds to a manageable few for testing. Rigorous validation, receptor preparation, and integration of AI tools are essential for reliable and reproducible results.
Virtual screening (VS) is defined as a computational method that automatically evaluates large libraries of small molecules against a biological target to identify the most promising candidates for experimental follow-up. Rather than synthesizing and testing thousands of compounds in the lab, researchers use VS to filter large chemical libraries down to a tractable set of high-confidence hits. This approach directly reduces both the time and cost of early drug discovery. Tools like molecular docking, pharmacophore modeling, and molecular dynamics simulations each play a defined role in this process. Platforms from NVIDIA and open-source pipelines on GitHub have made high-throughput VS accessible at a scale that was impractical a decade ago.
What are the main types of virtual screening methods?
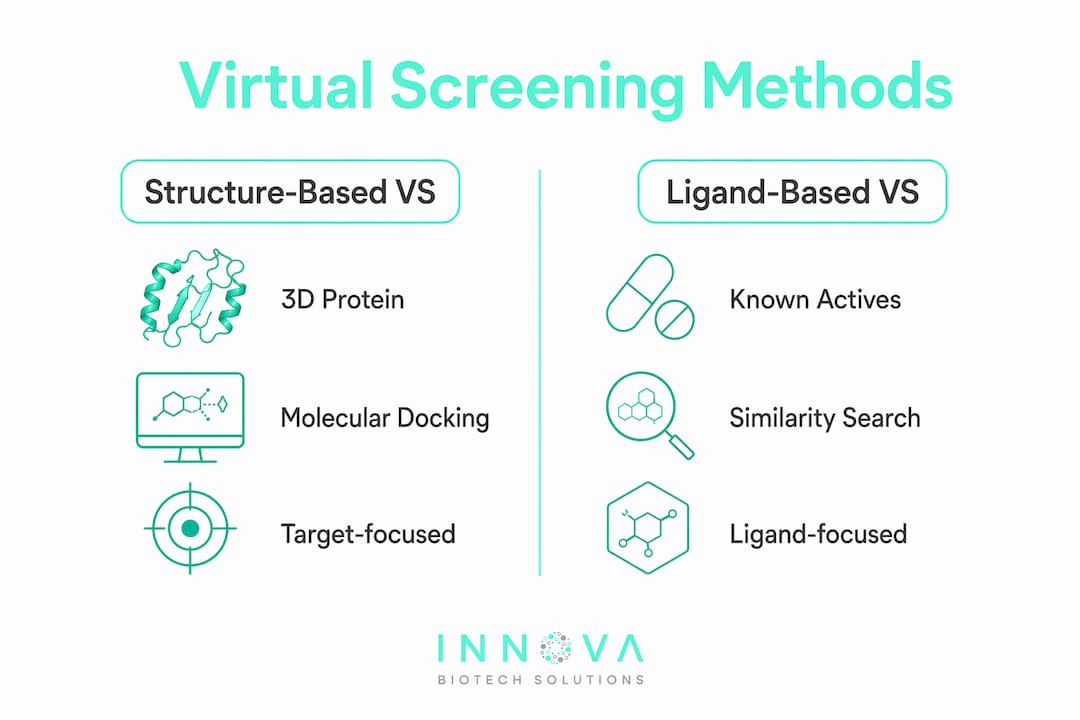
The two principal categories of virtual screening are structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS). They differ fundamentally in what information they require and how they rank compounds.
Structure-based virtual screening
Structure-based virtual screening uses the three-dimensional structure of the target protein, typically obtained through X-ray crystallography or cryo-EM, to simulate how candidate ligands bind within the active site. SBVS relies on molecular docking and scoring functions to predict binding poses and estimate affinity. This method is most powerful when a high-resolution target structure is available and the binding site is well-defined. Flexible receptor docking and ensemble approaches extend SBVS to targets with known conformational variability.

Ligand-based virtual screening
Ligand-based virtual screening takes a different starting point. It uses known active compounds as templates and searches for new molecules with similar chemical features, shapes, or pharmacophore patterns. LBVS is the method of choice when no experimental target structure exists. Pharmacophore modeling and shape-based similarity searches are the most common LBVS techniques. Filtering by pharmacophore or shape eliminates unlikely hits early, though accuracy depends heavily on conformer coverage and how well the template captures the binding pharmacophore.
SBVS vs. LBVS: a direct comparison
| Feature | Structure-Based VS | Ligand-Based VS |
|---|---|---|
| Primary input | 3D target protein structure | Known active ligands |
| Core method | Molecular docking, scoring functions | Pharmacophore modeling, shape similarity |
| Data requirement | High-resolution structural data | At least one confirmed active compound |
| Best use case | Well-characterized targets with solved structures | Targets lacking structural data |
| Key limitation | Sensitive to receptor preparation quality | Limited by template compound quality |
Neither approach is universally superior. Many modern campaigns run both in parallel and merge their ranked lists to capture hits that either method alone would miss.
How does a virtual screening workflow reduce thousands of compounds to dozens?
A well-designed virtual drug screening process operates as a sequential funnel. Each stage eliminates compounds that fail defined criteria, so only the most credible candidates reach experimental testing. Multi-stage funnel workflows routinely reduce a starting library of thousands of compounds to fewer than 50 for synthesis and assay.
Here is a representative workflow for a 1,000-compound library:
- Drug-likeness and physicochemical filtering. Apply Lipinski's Rule of Five, molecular weight cutoffs, and rotatable bond limits. A library of 1,000 compounds typically drops to 600–700 after this step.
- Pharmacophore or shape pre-filtering. Screen remaining compounds against a pharmacophore model or 3D shape query derived from a known active. This step can cut the pool to 200–300 compounds.
- Molecular docking and pose prediction. Dock filtered compounds into the target binding site. Score and rank by predicted binding energy. The top 10–15% advance, leaving roughly 20–45 compounds.
- Consensus rescoring. Apply two or more independent scoring functions to the docked poses. Consensus approaches reduce false positives by requiring agreement across models before a compound advances.
- ADMET and safety prediction. Filter for predicted absorption, distribution, metabolism, excretion, and toxicity liabilities using tools like SwissADME or pkCSM. Compounds with flagged liabilities are deprioritized.
- Final candidate selection. Manually inspect binding poses for interaction plausibility. Deliver 10–50 compounds to the chemistry team for synthesis and experimental validation.
Pro Tip: Build your pipeline as modular scripts or containerized steps from the start. Modular, standardized workflows make it straightforward to swap docking engines, update scoring functions, or reproduce results on a different computing environment without rebuilding from scratch.
The funnel structure is not just about efficiency. Each stage adds a different type of chemical intelligence, so the final shortlist reflects multiple orthogonal filters rather than a single scoring artifact.
How do you validate virtual screening results reliably?
Validation is where most virtual screening campaigns either earn or lose credibility. A high docking score alone does not confirm a true binder. Robust docking validation requires cross-docking, decoy benchmarking, and ROC analysis to assess whether the protocol genuinely enriches true actives over random compounds.
The core validation practices every researcher should apply:
- Pose recovery testing. Re-dock a co-crystallized ligand and measure the RMSD between the predicted and experimental pose. An RMSD below 2.0 Å is the standard threshold for acceptable pose accuracy.
- Cross-docking. Dock known actives into receptor conformations derived from different co-crystal structures. This tests whether the protocol generalizes beyond a single receptor snapshot.
- Decoy benchmarking. Prepare a set of property-matched decoys using tools like DUD-E or DEKOIS 2.0. Mix them with known actives and measure how well the docking protocol separates the two groups.
- AUC-ROC and enrichment factor. Calculate the area under the receiver operating characteristic curve and the enrichment factor at 1% and 5% of the ranked list. These metrics quantify how much better the protocol performs than random selection.
- Interaction plausibility check. Confirm that top-ranked poses form chemically sensible contacts with key residues. A pose that misses a catalytic residue or a known hydrogen bond acceptor is a red flag regardless of its score.
AI-driven docking methods still require these same validation steps. AI augments speed and sometimes accuracy, but it does not eliminate the need for rigorous benchmarking. Transparent reporting of all docking parameters, scoring functions, and validation metrics is what makes a VS campaign reproducible and publishable.
Pro Tip: Log every parameter change during protocol development, including receptor preparation steps, grid box dimensions, and scoring weights. Refer to 2026 validation best practices to align your reporting with current peer-reviewed standards.
How is virtual screening applied in modern drug discovery pipelines?
Virtual screening no longer operates as a standalone step. Integrative computational chemistry approaches now combine docking, pharmacophore modeling, molecular dynamics (MD) simulations, and AI-based scoring within a single discovery pipeline. Each method contributes a different layer of information, and their combination improves hit quality beyond what any single technique delivers.
Ensemble docking addresses one of the most persistent limitations of classical SBVS: protein flexibility. By docking against multiple receptor conformations derived from MD trajectories or crystallographic ensembles, researchers capture binding-competent states that a single rigid structure would miss. Multiple receptor conformations strongly influence enrichment rates and reduce false negatives in flexible targets like kinases and GPCRs.
AI and deep learning now serve as augmenting layers within these pipelines. AI-guided docking improves hit identification when integrated with physics-based validation strategies, but it does not replace them. Models trained on binding affinity data can rescore docked poses faster than traditional force fields, and graph neural networks are increasingly used to predict ADMET properties with higher accuracy than rule-based filters. For peptide-based drug candidates, deep learning augments physics-based screening by predicting binding affinity from sequence and structural features simultaneously.
| Computational Method | Primary Role | Key Strength |
|---|---|---|
| Molecular docking | Pose prediction and initial ranking | Speed across large compound libraries |
| Pharmacophore modeling | Feature-based pre-filtering | Captures key interaction geometry |
| Molecular dynamics | Conformational sampling and stability | Accounts for receptor and ligand flexibility |
| AI/deep learning rescoring | Binding affinity refinement | Learns nonlinear structure-activity patterns |
| ADMET prediction | Safety and drugability filtering | Flags liabilities before synthesis |
The practical impact is measurable. Integrating these methods accelerates lead identification and reduces the number of compounds that fail in later, more expensive experimental stages. The role of structural bioinformatics in connecting computational predictions to medicinal chemistry decisions has grown substantially as pipeline integration has matured.
Key takeaways
Virtual screening is most effective when structure-based and ligand-based methods are combined, validated with decoy benchmarking and AUC-ROC metrics, and integrated with ADMET filtering and AI rescoring within a modular, reproducible pipeline.
| Point | Details |
|---|---|
| Two core method types | SBVS uses 3D target structures; LBVS uses known active compounds as templates. |
| Funnel workflow design | Sequential filtering reduces thousands of compounds to 10–50 candidates for synthesis. |
| Validation is non-negotiable | Cross-docking, decoy sets, and AUC-ROC metrics confirm protocol reliability before synthesis. |
| Consensus scoring reduces noise | Combining multiple scoring functions lowers false positive rates across any docking campaign. |
| AI augments, not replaces | Deep learning rescoring improves hit quality only when paired with rigorous physics-based validation. |
Why receptor preparation is the step most researchers underestimate
After working through dozens of virtual screening campaigns, the single most consistent source of failure I see is not the choice of docking engine or scoring function. It is receptor preparation. Researchers spend weeks selecting compound libraries and tuning docking parameters, then spend 20 minutes preparing the receptor. That imbalance shows up directly in enrichment rates.
Protein structures from the Protein Data Bank often contain missing loops, alternate conformations, and crystallographic waters that need deliberate decisions before docking. Using a single receptor conformation for a flexible target like a kinase or an allosteric enzyme is a protocol choice that will suppress true positives regardless of how good the scoring function is. Ensemble docking is not a luxury for well-funded projects. It is a baseline requirement for any target with known conformational variability.
The second thing I would push back on is the assumption that a high AUC-ROC score during retrospective validation guarantees prospective success. Decoy sets can be biased toward easy separations if they are not carefully matched to the actives by molecular weight, logP, and rotatable bonds. I have seen campaigns with AUC values above 0.85 that still returned a majority of false positives in experimental testing, because the decoy set did not reflect the actual chemical space of the screening library.
My practical advice: treat validation as a design step, not a reporting step. Build your decoy set before you run your first docking job, and use it to make protocol decisions, not just to justify them after the fact.
— Hooman
How Innovabiotech supports your virtual screening projects
Innovabiotech brings together computational biology, bioinformatics, and medicinal chemistry expertise to support drug discovery teams at every stage of a virtual screening campaign.

From initial compound library curation and receptor preparation through docking execution, consensus rescoring, and ADMET filtering, Innovabiotech delivers fully customized pipelines built around your specific target and project goals. The team's peptide design and optimization services extend virtual screening results into de novo peptide candidates with predicted binding affinity and optimized pharmacological properties. For projects requiring protein engineering or enzyme modeling to complement computational screening, Innovabiotech's protein engineering services provide the structural and computational support needed to move from hit to lead with confidence. Contact Innovabiotech to discuss a tailored approach for your discovery program.
FAQ
What is virtual screening in drug discovery?
Virtual screening is a computational technique that evaluates large libraries of small molecules against a biological target to identify the most likely binders for experimental follow-up. It replaces or precedes high-throughput experimental screening to reduce cost and time in early drug discovery.
What is the difference between SBVS and LBVS?
Structure-based virtual screening uses the 3D protein target structure and molecular docking to rank compounds, while ligand-based virtual screening uses known active compounds as templates for pharmacophore or shape similarity searches. SBVS requires a solved target structure; LBVS requires at least one confirmed active ligand.
How many compounds does virtual screening typically evaluate?
Virtual screening campaigns commonly start with libraries ranging from thousands to millions of compounds and reduce them to 10–50 candidates through sequential filtering, docking, rescoring, and ADMET prediction before experimental testing.
What metrics are used to validate docking results?
The standard validation metrics are AUC-ROC, enrichment factor at 1% and 5% of the ranked list, and pose RMSD for re-docking known co-crystallized ligands. Cross-docking against multiple receptor conformations and decoy benchmarking provide additional confidence in protocol reliability.
Does AI replace traditional docking in virtual screening?
AI-guided docking augments traditional physics-based methods by improving rescoring speed and accuracy, but it does not replace rigorous validation workflows. Transparent, reproducible protocols remain required for confident hit identification regardless of the scoring method used.